Trajectory SP469
Force field:
martini_v2.2P
Simulation length (ns): 5000
Electric field (kJ mol-1 nm-1 e-1): 0
Temperature (K): 300 (v-rescale)
Pressure (bar): 1 (parrinello-rahman semiisotropic)
Number of particles: 24149
Time step (fs) : 25
Software: GROMACS 2020
Simulation length (ns): 5000
Electric field (kJ mol-1 nm-1 e-1): 0
Temperature (K): 300 (v-rescale)
Pressure (bar): 1 (parrinello-rahman semiisotropic)
Number of particles: 24149
Time step (fs) : 25
Software: GROMACS 2020
Supercomputer:
Finisterrae II CESGA
Peptides: P79 DRAMP02521
Lipids: DOPC, DOPE, DOPS, DPSM
Heteromolecules: CHOL
Ions: NA
Water model: PW
Peptides: P79 DRAMP02521
Lipids: DOPC, DOPE, DOPS, DPSM
Heteromolecules: CHOL
Ions: NA
Water model: PW
Sequence :
KFFKKLKNSVKKRAKKFFKKPRVIGVSIPF
Total charge (e): +12
Number of residues: 30
By amino acid: Basic: 12 Acidic: 0 Hydrophobic: 15 Polar: 3 Electrostatic Dipolar Moment (e nm): 12.1
Longitudinal (e nm): 12 Transversal (e nm): 1.89 Hydrophobic Dipolar Moment (nm): 38.2
Longitudinal (nm): 37.9 Transversal (nm): 4.7 Secondary structure: Helix
Activity:
Download Files
ITP file. JSON file. PDB file.
Click on any component to highlight it from the plot.
Upper leaflet
Lower leaflet
Lipids
Membrane model for: DOPC:DOPE:DOPS:DPSM:CHOL (3:3:3:2:4) (Cancer)
DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
Total charge (e): 0
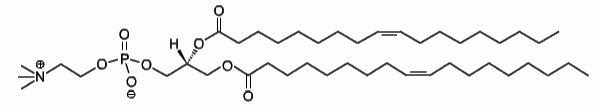
See DOPC lipid
Download ITP File. Download PDB File.
DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
Total charge (e): 0
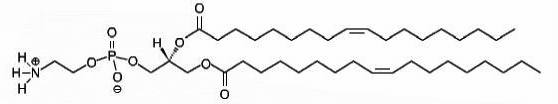
See DOPE lipid
Download ITP File. Download PDB File.
DOPS
1,2-dioleoyl-sn-glycero-3-phospho-L-serine
Total charge (e): -1
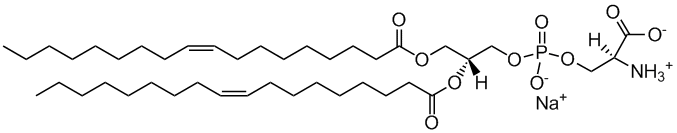
See DOPS lipid
Download ITP File. Download PDB File.
DPSM
Sphingomyelin C(d18:1/18:0) N-stearoyl-D-erythro tails
Total charge (e): 0
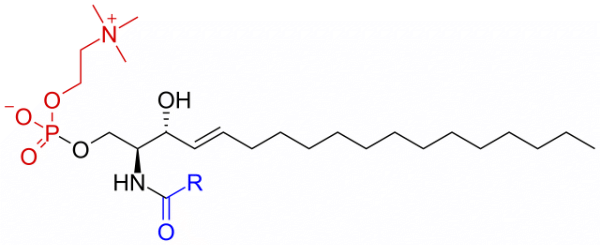
See DPSM lipid
Download ITP File. Download PDB File.
Last snapshot
Total contacts per residue
Molecular Dynamics based descriptors
Average and standard deviation,
calculated using the autocorrelation function (for time
series)
or the width of the distribution, for the last microsecond
of
the trajectory
Area per lipid
Membrane (nm2): 0.74878 ± 0.00074
Upper leaflet (nm2): 0.74878 ± 0.00074
Lower leaflet (nm2): 0.74878 ± 0.00074
Average Z coordinate
Peptide (nm): 6.382 ± 0.021
First Residue (nm): 6.418 ± 0.033
Last Residue (nm): 6.395 ± 0.035
Membrane (nm): 4.5856 ± 0.0045
Upper leaflet Head Group (nm): 6.5850 ± 0.0057
Lower leaflet Head Group (nm): 2.5939 ± 0.0032
Bilayer Thickness (nm): 3.9911 ± 0.0065
Peptide insertion (nm): -0.203 ± 0.022
Contacts
Peptide - Water: 59.81 ± 0.58
Peptide - Head groups: 17.48 ± 0.23
Peptide - Tail groups: 15.72 ± 0.27
Tilt (°): 90.93 ± 0.65
Membrane (nm2): 0.74878 ± 0.00074
Upper leaflet (nm2): 0.74878 ± 0.00074
Lower leaflet (nm2): 0.74878 ± 0.00074
Average Z coordinate
Peptide (nm): 6.382 ± 0.021
First Residue (nm): 6.418 ± 0.033
Last Residue (nm): 6.395 ± 0.035
Membrane (nm): 4.5856 ± 0.0045
Upper leaflet Head Group (nm): 6.5850 ± 0.0057
Lower leaflet Head Group (nm): 2.5939 ± 0.0032
Bilayer Thickness (nm): 3.9911 ± 0.0065
Peptide insertion (nm): -0.203 ± 0.022
Contacts
Peptide - Water: 59.81 ± 0.58
Peptide - Head groups: 17.48 ± 0.23
Peptide - Tail groups: 15.72 ± 0.27
Tilt (°): 90.93 ± 0.65
PepDF:
5(ns): CVS
Displacement (nm): 0.378 ± 0.019
Precession(°): 0.2 ± 1.0
50(ns) CVS
Displacement (nm): 1.07 ± 0.14
Precession(°): 1.8 ± 7.6
100(ns) CVS
Displacement(nm): 1.52 ± 0.48
Precession(°): 4.0 ± 14.0
200(ns) CVS
Displacement(nm): 2.3 ± 1.3
Precession(°): 9.0 ± 26.0
Download JSON File.
5(ns): CVS
Displacement (nm): 0.378 ± 0.019
Precession(°): 0.2 ± 1.0
50(ns) CVS
Displacement (nm): 1.07 ± 0.14
Precession(°): 1.8 ± 7.6
100(ns) CVS
Displacement(nm): 1.52 ± 0.48
Precession(°): 4.0 ± 14.0
200(ns) CVS
Displacement(nm): 2.3 ± 1.3
Precession(°): 9.0 ± 26.0
Download JSON File.
Peptide Analyses
Peptide Displacement Fingerprint
(PepDF)
Lateral displacement vs
Rotational
Displacement along the trajectory, for different time
windows .
Density maps:
2D-density maps of lipids around the
peptide
along XY and YZ axis, calculated for each lipid type along the
last
microsecond.
Lipid-Peptide Analyses:
z-Position
Z-coordinate, averaged for
differetn
parts of the the system: peptide, membrane, first and
last
backbone (BB) residues and upper of lower leaflet
lipids’
headgroups (HGs).
Minimum distance
Minimum distance (nm) between the
peptide backbone and the lipids (headgroups and
tailgroups).
Number of contacts
Number of contacts between the
peptide backbone and the water or the lipids separated
by
lipid headgroups (HG) or lipid tails, using a cut-off of
0.6
nm.
Lateral density
Lateral density for the different
components of the system: headgroups, tail groups,
peptide
and water.